Why popEVE Matters
Interpreting missense variants has long been one of the most persistent challenges in clinical genetics. These single–amino-acid substitutions can subtly alter protein structure or function, yet their effects are often context-dependent and difficult to classify reliably. Existing prediction tools frequently perform well within known disease genes but struggle to offer consistent, proteome-wide calibration. This gap limits their diagnostic utility, particularly for rare disorders where clinicians often face variants that have never been observed before.
A new study in Nature Genetics introduces popEVE, an AI model that represents a substantive advance in this field. Developed by researchers at the Centre for Genomic Regulation in Barcelona and Harvard Medical School, popEVE integrates deep evolutionary information with large-scale human population data to estimate variant severity across the entire proteome. The objective is to provide a continuously calibrated score for each variant—one that reflects biological impact and can be meaningfully compared across genes.
The scientific rationale is strong. Evolutionary sequence diversity across hundreds of thousands of species captures which amino-acid substitutions biology has historically tolerated. Human population datasets such as UK Biobank and gnomAD reveal which variants are compatible with healthy human physiology today. popEVE fuses these signals through a generative probabilistic framework incorporating a large-language protein model and Gaussian process calibration. This combined approach allows the model to distinguish pathogenic from benign variants, assess severity gradients, and avoid the common problem of overpredicting pathogenicity in healthy individuals.
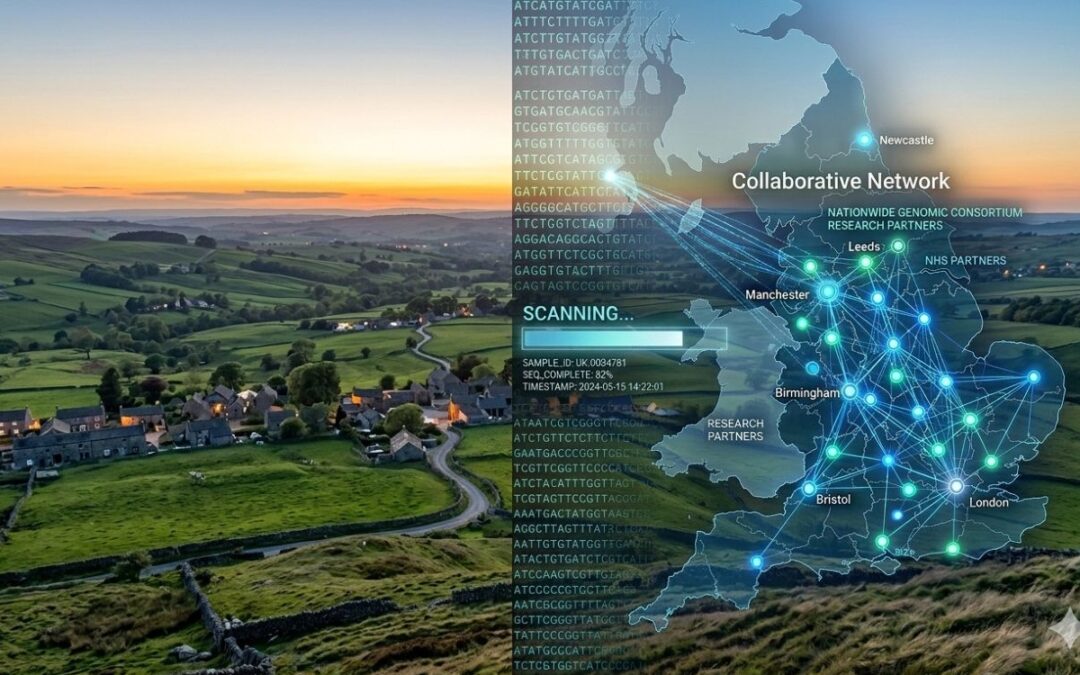
The model’s performance on real-world data is noteworthy. In a cohort of 31,000 families affected by severe developmental disorders, popEVE correctly identified the most damaging de novo missense variant in 98 per cent of cases. This capability is particularly impactful in singleton sequencing, where only the child’s genome is available. The model’s ability to infer whether a variant is likely de novo—even without parental DNA—has important implications for clinical practice worldwide, especially in settings where trio sequencing is impractical.

popEVE also expands the landscape of genes implicated in developmental disorders. In the study, the model flagged variants in 442 genes, including 123 not previously associated with such conditions. These candidates show hallmark features of disease genes, including neurodevelopmental expression profiles, essential functions, and participation in known protein interaction networks. Structural analyses further indicate that predicted deleterious variants often localize to functionally critical protein interfaces, providing mechanistic support for their involvement.
Harvard Medical School’s summary underscores additional translational dimensions. popEVE successfully distinguishes childhood-lethal variants from those manifesting later in life, detects whether alterations were inherited or arose de novo, and shows no ancestry bias. The model’s integration into portals such as ProtVar and UniProt will enable global use, while early clinical testing—ranging from Europe to Senegal—has already helped resolve previously undiagnosed cases. The team highlights its potential not only for diagnosis but also for identifying new therapeutic targets, as prioritizing variants by predicted severity can reveal upstream pathways suitable for drug discovery.
On key benchmarks, popEVE outperforms existing tools such as DeepMind’s AlphaMissense, especially in distinguishing severe early-onset pathogenic variants from milder ones. It is computationally efficient, allowing for broader deployment in low- and middle-income countries, where resource constraints often limit genomic analysis.
Ultimately, what sets this work apart is its ability to unify variant interpretation through a calibrated, evolution-informed scoring system that applies to every human gene. Missense interpretation has long been limited by inconsistent calibration and reliance on prior knowledge of disease genes. popEVE offers a more general framework: one that can prioritise variants, identify novel disease candidates, support clinicians in challenging cases, and—crucially—bring undiagnosed patients closer to answers.
For the global rare-disease community, the implications are significant. An estimated 300–400 million people worldwide are affected by rare disorders, many of whom endure years-long diagnostic odysseys. While no model can guarantee a diagnosis, popEVE represents a meaningful advance toward more precise, scalable, and equitable genomic interpretation. It brings the field a step closer to realising the full promise of clinical sequencing and to identifying new targets for therapeutic innovation.
© Dr. Robert Siegmund, Life Code GmbH, Switzerland.
www.lifecode.ch